Transform H5AD for faster access
Conver to to gene-major using AnnData in python script
Developed by Gabriel Hoffman
Run on 2026-05-19 13:56:48
Source:vignettes/h5ad.Rmd
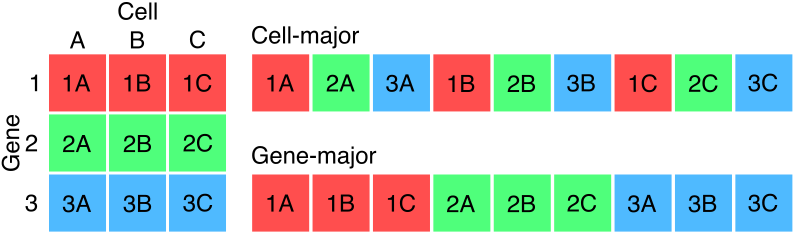
h5ad.RmdCell- versus gene-major storage
General matrices can be stored either in row-major or column-major format, depending on the need for efficient access to rows versus columns. Similarly, sparse matrices with many zero entries can be stored in CSR or CSC format. In both cases, the best format depends on the need to access rows versus columns most efficiently.
The H5AD format from AnnData is widely used to store single cell gene expression data. Many standard analyses are cell-centric tasks: QC, annotation, PCA, UMAP, K-Nearest Neighbors. Therefore, by default, an H5AD file stores the sparse read count matrix in a format that prioritizes efficient access to cells rather than genes. Avoiding the details of the underlying data storage model, we term this a cell-major format. This is the default format used by most H5AD files.
However, differential expression analysis is a gene-centric task. For
each annotated cell type, lucida fits a negative binomial
regression model to each gene. Therefore, lucida analysis
can be accelerated by storing the read count matrix in a
gene-major format (Figure 1) also supported by
H5AD.
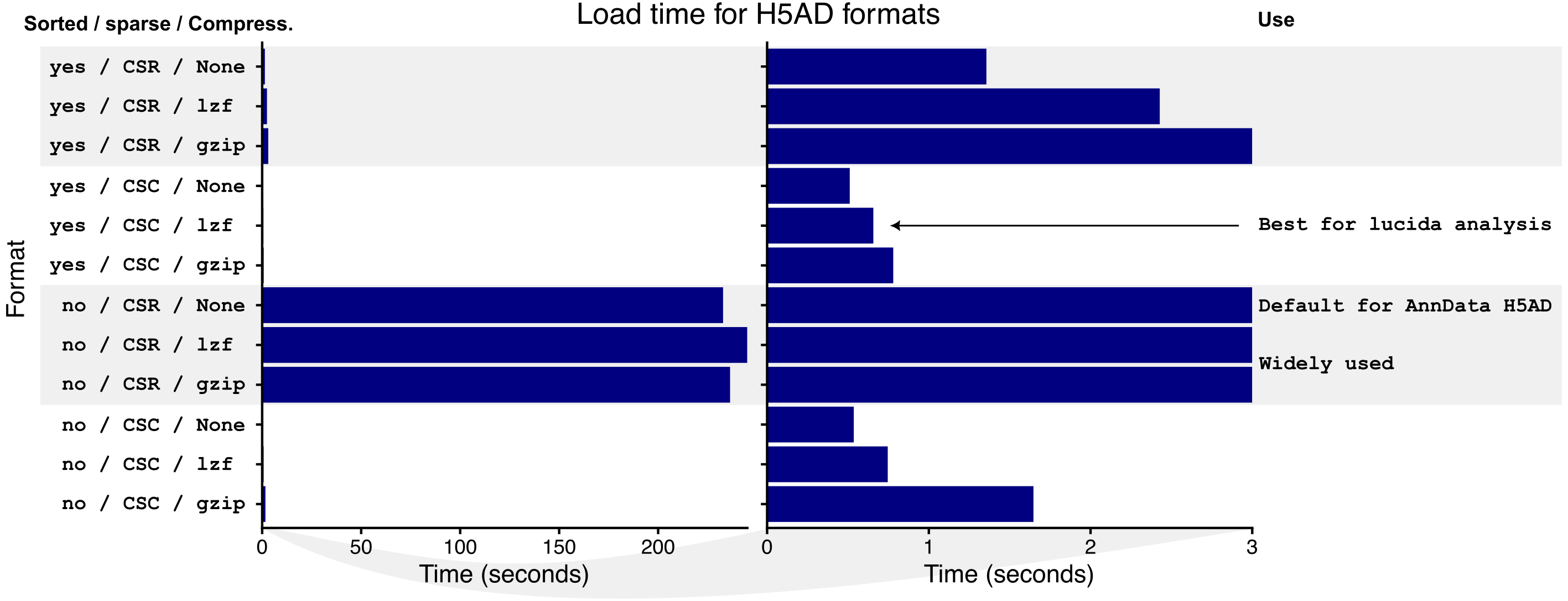
Empirical testing of H5AD formats
Load times for 12 H5AD formats were benchmarked and show that using a gene-major format can outperform the default cell-major format (Figure 2). Best performance was observed here and in other datasets using gene-major format, compression using the LZF algorithm, and sorting cells by annotated class and biological sample identifier.
Convert H5AD file
We developed the recode_h5ad.py script that uses the AnnData library in order to:
- Convert read count matrix to gene-major format
- Sort cells by annotated class and biological sample identifier
- Create variable
libSizestoring the number of reads for each cell. Ifraw/Xexists, computed from this. Otherwise computed fromX - Write to new H5AD file using LZF compression
The result is a valid H5AD file supported by standard tools, but just optimized for gene-centric access.
Here, we download the script and examine the arguments:
# Download python script
SRC=https://raw.githubusercontent.com/GabrielHoffman/GenomicDataStream_analysis/refs/heads/main/recode_h5ad.py
wget $SRC
# Install anndata if needed
conda install anndata
# Examine arguments
recode_h5ad.py --help
usage: recode_h5ad.py [-h] --input INPUT --output OUTPUT [--ondisk]
[--sortBy SORTBY] [--compression {None,None,gzip,lzf}]
[--format {CSR,CSC}] [--noLibSize]
Convert an AnnData .h5ad file so that X (and raw/X) is stored in CSC sparse format (v1.1)
options:
-h, --help show this help message and exit
--input INPUT Input .h5ad file
--output OUTPUT Output .h5ad file
--ondisk Use file-backed mode to reduce memory usage
--sortBy SORTBY Cols to sort by in _decreasing_ order of importance
--compression {None,gzip,lzf}
Optional compression for output file (gzip or lzf).
Default: None
--format {CSR,CSC} Store sparse count matrix in CSR or CSC format. CSR
allows faster access to cells, CSC gives faster
access to genes. Default: CSR
--noLibSize Skip computing libSize for each cellNow, we convert an H5AD file to CSC (i.e. gene-major) format, sorting the cells by class, subclass and SampleID, and saving using LZF compression.
# H5AD=(Original H5AD file)
# OUTFILE=(New H5AD file)
recode_h5ad.py \
--input $H5AD \
--sortBy class,subclass,SampleID \
--format CSC \
--compression lzf \
--out $OUTFILESession info
## R version 4.5.1 (2025-06-13)
## Platform: aarch64-apple-darwin23.6.0
## Running under: macOS Sonoma 14.7.1
##
## Matrix products: default
## BLAS/LAPACK: /opt/homebrew/Cellar/openblas/0.3.31_1/lib/libopenblasp-r0.3.31.dylib; LAPACK version 3.12.0
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## time zone: America/New_York
## tzcode source: internal
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## loaded via a namespace (and not attached):
## [1] digest_0.6.39 desc_1.4.3 R6_2.6.1 fastmap_1.2.0
## [5] xfun_0.56 cachem_1.1.0 knitr_1.51 htmltools_0.5.9
## [9] rmarkdown_2.30 lifecycle_1.0.5 cli_3.6.5 sass_0.4.10
## [13] pkgdown_2.2.0 textshaping_1.0.5 jquerylib_0.1.4 systemfonts_1.3.2
## [17] compiler_4.5.1 tools_4.5.1 ragg_1.5.1 bslib_0.10.0
## [21] evaluate_1.0.5 yaml_2.3.12 otel_0.2.0 jsonlite_2.0.0
## [25] rlang_1.1.7 fs_1.6.7 htmlwidgets_1.6.4