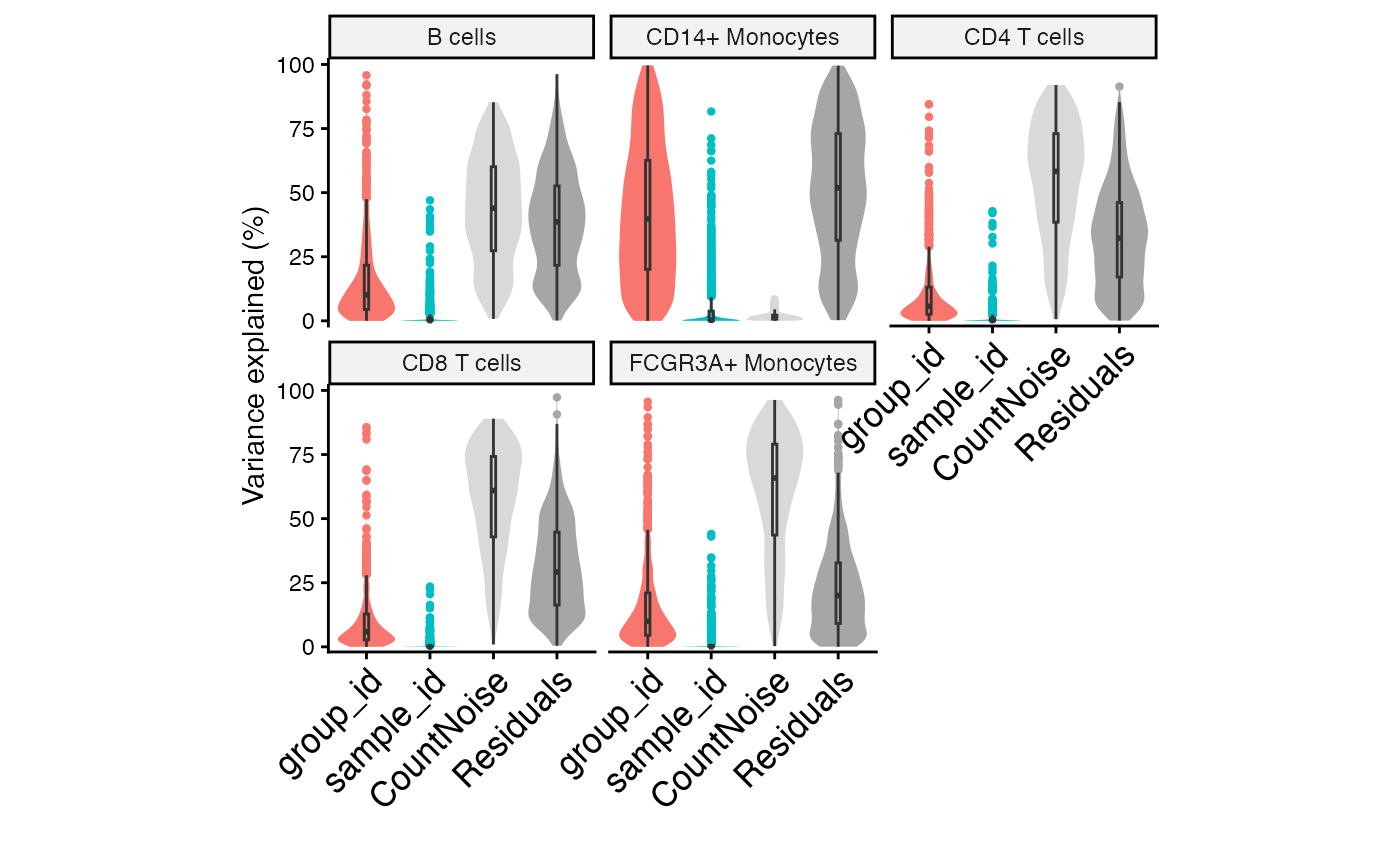
Variance partitioning analysis on each gene
Details
Using lambda.method = "parametric" requires refitting the model without the fixed effects. This is more accurate, especially for responses with low counts. Using lambda.method = "mean" is a fast approximation that works well for responses with sufficient counts. This methid can _under-estimate_ the count noise.
Examples
library(SingleCellExperiment)
# Load example data
data(example_sce, package="muscat")
sce <- example_sce
# Compute library size for each cell
sce$libSize <- colSums(counts(sce))
# Specify regression formula and cell annotation
form <- ~ group_id + (1|sample_id)
fit <- lucida(sce, form, "cluster_id", verbose=FALSE)
#> B cells
#> CD14+ Monocytes
#> CD4 T cells
#> CD8 T cells
#> FCGR3A+ Monocytes
# Model with only intercept and random effect
form <- ~ (1|sample_id)
fit.null <- lucida(sce, form, "cluster_id", verbose=FALSE)
#> B cells
#> CD14+ Monocytes
#> CD4 T cells
#> CD8 T cells
#> FCGR3A+ Monocytes
# Variance partitioning analysis
vp <- fitVarPart(fit, fit.null)
plotVarPart(sortCols(vp))