Functional Interpretation of Genetic Variants Using Deep Learning Predicts Impact on Epigenome
Abstract
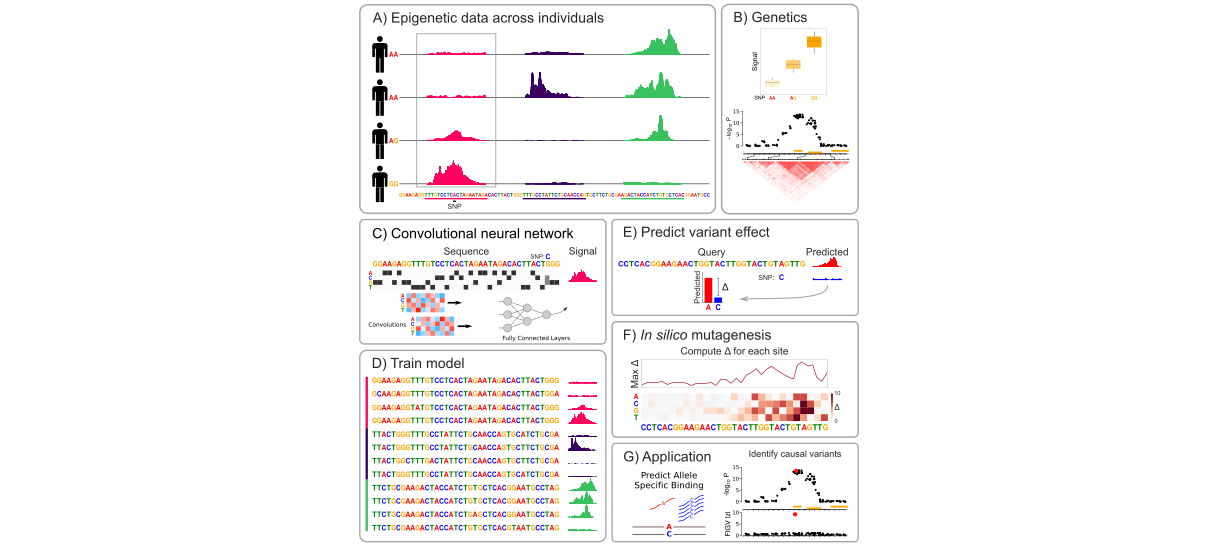
Identifying causal variants underling disease risk and adoption of personalized medicine are currently limited by the challenge of interpreting the functional consequences of genetic variants. Predicting the functional effects of disease-associated protein-coding variants is increasingly routine. Yet the vast majority of risk variants are non-coding, and predicting the functional consequence and prioritizing variants for functional validation remains a major challenge. Here we develop a deep learning model to accurately predict locus-specific signals from four epigenetic assays using only DNA sequence as input. Given the predicted epigenetic signal from DNA sequence for the reference and alternative alleles at a given locus, we generate a score of the predicted epigenetic consequences for 438 million variants. These impact scores are assay-specific, are predictive of allele-specific transcription factor binding and are enriched for variants associated with gene expression and disease risk. Nucleotide-level functional consequence scores for non-coding variants can refine the mechanism of known causal variants, identify novel risk variants and prioritize downstream experiments.